
次世代のエネルギー貯蔵デバイスとして期待を集める全固体電池(All-Solid-State Batteries; ASSB)。その心臓部とも言える固体電解質の研究において、人工知能(AI)を用いた原子レベルのシミュレーションが、これまでの「常識」を覆す決定的な知見をもたらした。
韓国科学技術研究院(KIST)のByungju Lee博士率いる研究チームは、アモルファスリチウム・リン・硫黄(Li-P-S; LPS)系電解質におけるイオン輸送メカニズムを、AIベースの計算手法によって精密に解析 。その結果、リチウムイオンの伝導を支配するのは、経路の「つながり」よりも、局所的な「移動のしやすさ」であることを突き止めたのだ。
全固体電池が直面する「アモルファス」というブラックボックス
現在のリチウムイオン電池は液体電解質を使用しているが、これは外部からの衝撃や過熱によって発火・爆発するリスクを孕んでいる。この安全性の課題を解決するのが、不燃性の固体電解質を用いる全固体電池である 。
数ある固体電解質候補の中でも、アモルファス硫化物電解質は、以下の特性から極めて有望視されている。
- 機械的柔軟性: 固体同士の界面形成が容易で、加工性に優れる 。
- 等方的なイオン輸送: 結晶構造のような特定の方向に依存しない伝導が可能である 。
- 粒界抵抗の低減: 結晶粒界が存在しないため、抵抗を抑制できる 。
しかし、アモルファス材料には「長距離秩序(Long-range order)」が存在しない。原子が不規則に並んでいるため、従来の結晶学的なモデルではイオンがどのように移動しているのかを正確に把握することが困難だった 。これまで、電解質の性能向上は、経験的な組成調整や加圧条件の変更といった試行錯誤に頼る部分が大きく、性能差が生じる根本原因を体系的に説明する枠組みが欠如していたのである 。
AIシミュレーション:原子のダンスを可視化する
KISTの研究チームは、この「解析の限界」を突破するために、機械学習ベースの分子動力学(Molecular Dynamics; MD)シミュレーションを導入した 。
従来手法の限界とAIの優位性
従来の第一原理計算(DFT)は、量子力学に基づいた高い精度を持つが、計算コストが膨大である。そのため、数千個の原子を含むアモルファス構造や、長時間の拡散現象をシミュレートするには限界があった 。
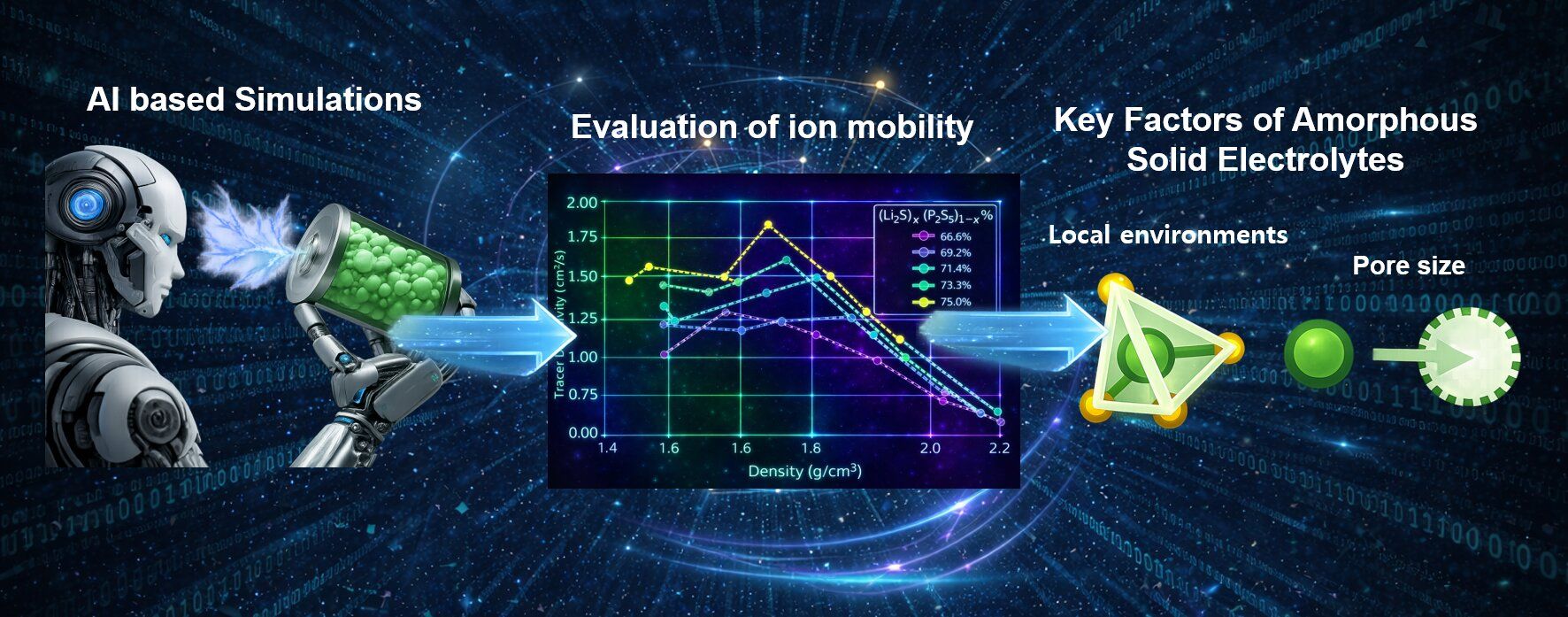
研究チームは、DFTで得られた膨大なデータセットを学習させた「ニューラルネットワークポテンシャル(Neural Network Potentials; NNPs)」を構築した 。これにより、DFTに近い精度を維持しつつ、従来よりも数千倍高速に大規模な原子挙動を解析することが可能となった。具体的には、1000個以上の原子を含むシミュレーションセルを用い、様々な組成と密度条件下でのリチウムイオンの動きを1ナノ秒(\(10^{-9}\)秒)にわたって追跡した 。
構造生成の新たなアプローチ
研究チームは、従来の「融液急冷法(Melt-quench method)」ではなく、ポリアニオンユニット(\(P_2S_7^{4-}\), \(PS_4^{3-}\), 孤立した \(S^{2-}\))と \(Li^+\) イオンをランダムに配置し、等温等圧(NPT)アンサンブルで平衡化させる手法を採用した 。これにより、特定のポリアニオン分布を系統的に制御し、組成と密度が拡散に与える影響を独立して評価することに成功した 。
常識を覆す発見:伝導を支配するのは「短距離の跳躍」
本研究の最大の成果は、リチウムイオンの拡散を「短距離の跳躍(Short-range hopping)」と「長距離の経路接続性(Long-range connectivity)」の2つの要素に分解して評価した点にある 。
拡散定数の分解式
トレーサー拡散係数 \(D_{tracer}\)は、以下の式で表される。
\(D_{tracer} = f \times D_{hop}\)
ここで、\(D_{hop}\)は原子間での局所的な跳躍のしやすさを示す「跳躍拡散係数」、\(f\) は移動経路の直線性を表す「自己相関因子(Correlation factor)」である 。
解析の結果、イオン伝導性能(拡散係数)の変動幅において、\(D_{hop}\)は組成や密度によって最大5倍の差が生じるのに対し、\(f\) の変動は2倍程度に留まることが判明した 。これは、アモルファスLPS電解質において、イオンがネットワーク全体をどう通り抜けるか(経路のつながり)よりも、「隣のサイトへ移動できるか」という局所的なダイナミクスこそが、全体の性能を決定づける支配的要因であることを示唆している 。
理想的な「巣」と「隙間」の特定
研究チームはさらに踏み込み、短距離跳躍を促進する具体的な構造条件を特定した。
\(Li-S_4\)配位環境の重要性
リチウムイオン(\(Li^+\))を取り囲む硫黄原子(\(S\))の数(配位数学)が重要である。研究によれば、リチウムが4つの硫黄原子に囲まれた \(Li-S_4\)四面体構造 の割合(\(\varphi_{LiS_4}\))が高いほど、跳躍拡散係数 \(D_{hop}\)が線形に向上することが明らかになった 。
この \(Li-S_4\)環境は、エネルギー的にリチウムを安定化させつつ、適切な歪み(Distortion)を伴うことでポテンシャルエネルギー曲面を平坦化し、イオンの離脱と移動を容易にする「理想的な巣」として機能する 。密度が高くなりすぎると配位数が5以上に増加し、逆にリチウムが強く拘束されてしまうため、拡散性能は低下する 。
ポア(空隙)進化のパラドックス
もう一つの決定的な要因は、構造内の「空隙(Pore)」のサイズである。これまでの材料開発では、「密度を下げて自由体積(Free volume)を増やせば、イオンは動きやすくなる」と考えられてきた。
しかし、AI解析はこの定説を修正した。
- 有効なチャンネル: 最大ポア径(\(d_{max}\))が \(6.0 \text{ \AA}\)未満 の場合、空隙はリチウムの移動経路を広げる「アクセス可能なチャンネル」として機能し、拡散を促進する 。
- 不活性な空洞: ポア径が \(6.0 \text{ \AA}\) を超える と、それはリチウムを安定化させるアニオンが存在しない「不活性な空洞」となり、リチウムを捕捉できないため、実質的な拡散ボリュームが減少して性能が低下する 。
このメカニズムにより、LPS系電解質においては密度 \(1.8 \text{ g cm}^{-3}\)付近、組成 \(Li_3PS_4\)付近で最適な導電率が得られることが理論的に裏付けられた 。
産業界へのインパクト:試行錯誤からの脱却
本研究が提示した「局所配位(化学的安定化)」と「ポア進化(物理的トポロジー)」という二元的なフレームワークは、今後の電池開発の在り方を根本から変える可能性がある 。
製造プロセスの最適化
電解質の組成比だけでなく、ペレット成形時の圧力や熱処理条件を調整することで、内部の \(Li-S_4\)配位率とポアサイズを制御し、追加の材料開発なしにイオン伝導性を最大限に引き出すことが可能になる 。これは、産業現場でのコスト削減と性能向上を直結させる知見である。
他材料への展開
このAIベースの解析手法は、LPS系だけでなく、酸化物系(\(Li_3PO_4\) など)や他のアモルファス固体電解質にも適用可能であることが示されている 。高パフォーマンスな候補材料を事前にAIで選別することで、材料開発のスピードは劇的に加速するだろう。
AIが拓く全固体電池の未来

KISTの研究チームによる今回の成果は、長年「不規則さ」ゆえに解明が遅れていた非晶質材料の伝導メカニズムに、明確な物理的指標を与えた。
「密度は低いほど良い」という思い込みを排し、原子レベルの「巣」の心地よさと「通り道」の広さをAIで精密に設計する。この新しいパラダイムは、電気自動車(EV)やエネルギー貯蔵システム(ESS)の安全性を飛躍的に高める全固体電池の商用化を、最短距離へと導くはずだ 。
AIと材料科学の融合は、もはや単なる補助ツールではなく、次世代エネルギーの扉をこじ開ける「鍵」そのものとなったのである。
論文
- Advanced Energy Materials: Origin of Optimal Composition and Density for Li-Ion Diffusion in Amorphous Li–P–S Electrolytes
参考文献


